Manual
PRODIGY (PROtein binDIng enerGY prediction) is a web application to predict the binding affinity of protein-protein complexes based on intermolecular contacts.
Input
- The protein-protein complex
- PRODIGY server takes as input the three-dimensional structure of the protein-protein complex in PDB or mmCIF format:
-
You can choose one of the following options:
- upload the 3D structure coordinates in PDB or mmCIF format - provide a protein databank ID code for automatic retrieval from the Protein Data Bank - Upload a multi-model PDB ensemble or .zip file with multiple structures to analyzing multiple structures at the same time
(i.e. models derived from docking simulations) - Specify the chains
-
The user is required to specify the chain identifiers for the molecules involved in the interaction.
Interacting molecules associated with multiple chain IDs should be provided as comma-separated list.
For the FAB/HIV-1 capsid protein p24 complex (PDB code 1E6J) the input would be expected in following format:Interactor 1: P Interactor 2: L,H In this example, the binding affinity will be predicted for the interface made between chain P (p24) and chains L and H (FAB).
- Please note! PRODIGY only supports the 20 standard amino acids
- Archive file
- When submitting an archive file to PRODIGY, make sure that the chain IDs of ALL structures are compatible with the Interactors specified in the submission form.
Parameters (optional)
- Temperature (in ℃)
- By default the value of the dissociation constant (Kd) is calculated at 25 ℃. The user can change this value to any desired temperature.
- Job ID
- The user can specify a personalized Job ID to identify the run.
- If an email is provided, a link with the results will be sent when the job is done. The results will be stored for 2 weeks.
Output
PRODIGY outputs are displayed online and remain downloadable for 14 days. A link to the online resource is also emailed to the user, if an email is provided.
The results returned by the server include:
| 1. | Predicted value of the binding affinity (ΔG) expressed in kcal mol-1 |
| 2. | Calculated value of the dissociation constant (Kd) at a given temperature (25 ℃ by default), expressed in Molar (M) |
| 3. | Number of intermolecular contacts (ICs) at the interface within the threshold distance of 5.5 Å, separately listed according to the contact property |
| 4. | Percentage of the charged and apolar non-interacting surface (NIS%) of the complex |
| 5. | Downloadable table (.txt) listing the residues in contact within the given threshold |
| 6. | Downloadable ready-to-run Pymol (Delano Scientific, 2002) script (.pml) with different color coding for the interacting residues |
| 7. | Archive file including all the output files |
How to run PRODIGY locally
In case you have many structures on which you wish to run PRODIGY, we distribute the standalone version in form
of a Github repository.
The link to the repository can be found at the BonvinLab software page.
PRODIGY(PROtein binDIng enerGY prediction - LIGands) is an automatic web server for the fast prediction of binding affinity of protein-small ligand (such as drugs or metabolites) complexes. Being a structural-based approach, it is suitable for every kind of protein-small ligand complex .
Input
- The protein-small ligand complex.
- PRODIGY server takes as input the three-dimensional structure of the protein-ligand complex in PDB or mmCIF format:
-
You can choose one of the following options:
- upload the 3D structure coordinates in PDB or mmCIF format - provide a protein databank ID code for automatic retrieval from the Protein Data Bank - Upload a multi-model PDB ensemble or .zip file with multiple structures to analyzing multiple structures at the same time
(i.e. models derived from docking simulations) - Specify the chains
-
The user is required to specify the chain identifiers for the molecules involved in the interaction, both for the Protein and the Ligand as well as the Residue identifier for the ligand. Example: in the complex of MDM2 with a pyrrolidine MDM2 inhibitor (PDB code 4JRG) the input would be expected in the following format:
Protein Chain ID: A Ligand ID: A:I09 In this example, the binding affinity will be predicted for the interaction between the protein and the ligand present in chain A.
Please note! PRODIGY-LIG only supports the 20 standard amino acids for the Protein Chain
When submitting an archive file to PRODIGY, make sure that the chain IDs of ALL structures are compatible with the Interactors specified in the submission form.
Parameters (optional)
- Electrostatic interaction
- You can provide the electrostatic interaction energy calculated by the HADDOCK refinement page. If you submit a PDB file obtained directly from the HADDOCK refinement page, PRODIGY-LIG will automatically retrieve the energy from it. Otherwise, the energy can be added manually in the "Electrostatic Energy" box.
-
If no electrostatic energy is provided , the server will run the "No electrostatics prediction"
protocol (see below and in "Methods").
- Check also the paragraph below "How do I calculate the electrostatic energy of the complex?"
- Job ID
- The user can specify a personalized Job ID to identify the run.
- If an email is provided, a link with the results will be sent when the job is done. The results will be stored for 2 weeks.
Output
PRODIGY-LIG outputs are displayed online and remain downloadable for 14 days. A link to the online resource is also emailed to the user, if an email is provided.
The results returned by the server include:
| 1. | Predicted value of the binding affinity (ΔG) expressed in kcal mol-1 |
| 2. | Number of intermolecular atomic contacts (ACs) at the interface between the protein and the ligand within the threshold distance of 10.5 Å. Contacts are divided by atom-type. |
| 3. | Downloadable table (.txt) listing the residues in contact within the given threshold |
| 4. | Archive file including all the output files |
How do I calculate the electrostatic energy of the complex?
To refine your protein-small ligand complex and obtain the electrostatic energy, go to the HADDOCK refinement web page. Make sure to register for a HADDOCK account in order to submit jobs, use the registration page). In this example, we will refine the protein-ligand complex with PDB code 4JRG.
Note: The blue bars on the server can be folded/unfolded by clicking on the arrow on the right
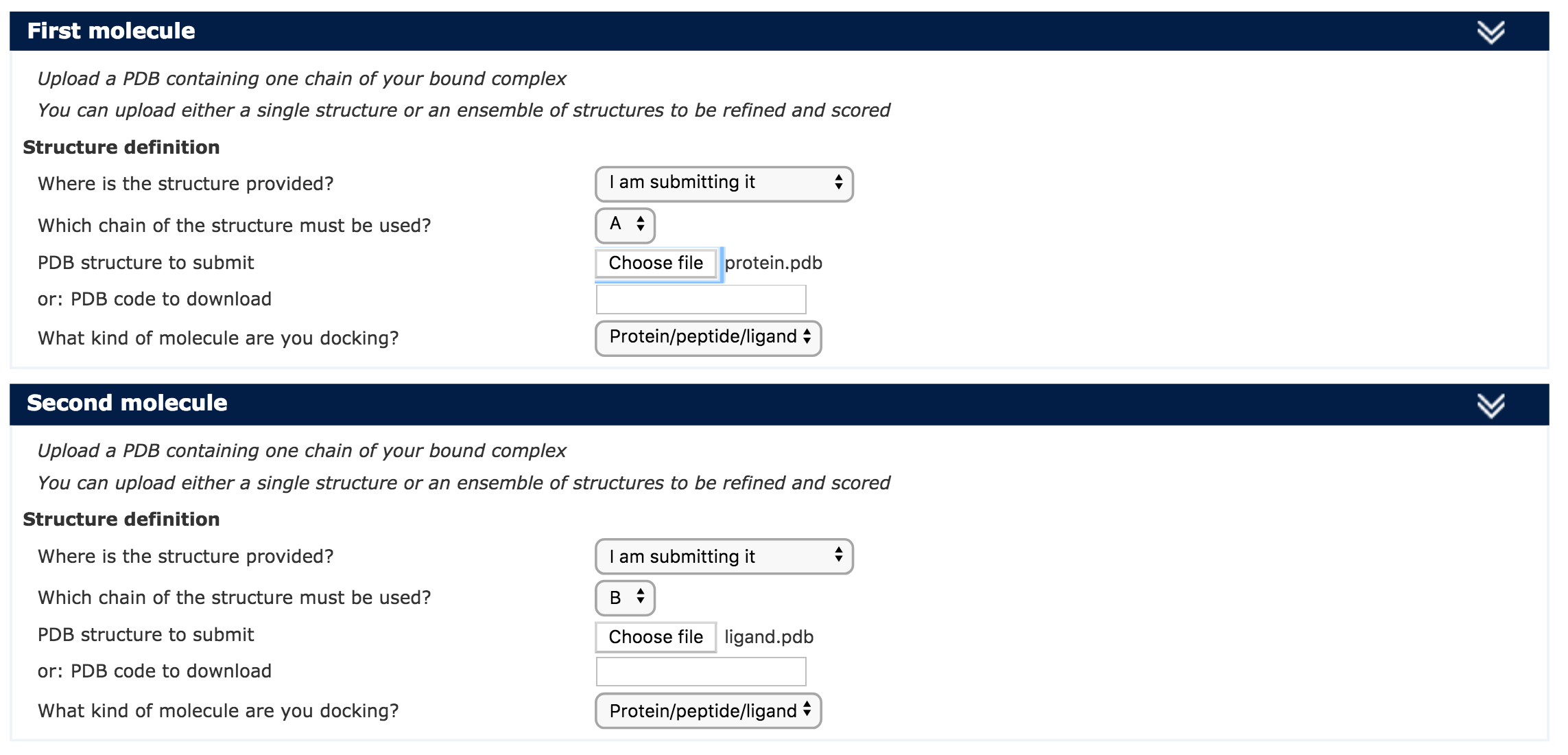
| Step 1 | Define a name for your docking run, i.e. 4JRG_refinement |
| Step 2 | Input the protein PDB file, or type the PDB code with the chain identifier. For this unfold the First Molecule menu. |
| Step 3 | Input the ligand PDB file, or type the PDB code with the chain identifier. For this unfold the Second Molecule menu. |
| Step 4 | You are ready to submit! |
Once your run has completed you will be presented with a result page showing the cluster statistics and some graphical representation of the data.
In the refinement, there will be only 1 cluster considering that the models represent just a refinement of the bound input structure. The various components of the HADDOCK score are reported on the result web page.

The Electrostatic energy (in this example -94.1) is the energy feature that you are looking for to provide at the PRODIGY-LIG page. Alternatively, you can also download the top refined model from the refinement page (click on Download next to "Nr 1 best structure") and upload it to PRODIGY-LIG. The information about the electrostatic energy is already contained in the file and will be automatically retrieved from our web-server.
Can I provide the electrostatic energy calculated by other tools than HADDOCK?
No, PRODIGY-LIG is an empirical approach that was trained by using the electrostatic interactions energy calculated by HADDOCK. The prediction formula has been therefore optimized according to this feature.
PRODIGY-CRYSTAL (PROtein binDIng enerGY prediction to classify CRYSTALlographic interfaces) is an automatic web server for distinguish crystallographic from biological interfaces in protein complexes. PRODIGY-CRYSTAL implements an easy and fast classification approach based on the type and nature of intramolecular contacts at the interface.
Input
- Classification of biological/crystallographic interfaces
- Specify the chains
- Please note! PRODIGY-CRYSTAL only supports the 20 standard amino acids and process binary complexes (two chains).
PRODIGY-CRYSTAL server takes as input the three-dimensional structure of the protein-protein complex in PDB or mmCIF format.
You can choose one of the following options:
| - | Upload the 3D structure coordinates in PDB or mmCIF format |
| - | Provide a protein databank ID code for automatic retrieval from the Protein Data Bank |
| - | Upload an archive file (.zip) or multi-model PDB ensemble for analysing multiple structures at the same time (i.e. models derived from docking simulations). When submitting an archive file to PRODIGY-CRYSTAL, make sure that the chain IDs of ALL structures are compatible with the chains specified in the submission form |
The user is required to specify the chain identifiers for the proteins involved, for which the nature of the interface is wished to be predicted.
Interacting molecules associated with multiple chain IDs should be provided as comma-separated list.
Example: in the complex between the bovine beta-trypsin and CMTI-I (PDB code 1PPE) the input would be expected in the following format:
| Interface 1 Chain ID: E |
| Interface 2 Chain ID: I |
In this example, the nature of the complex (biological or crystallographic) will be predicted for the interface between chain E and chain I.
Parameters (optional)
- Job ID
- The user can specify a personalized Job ID to be used instead of the randomly generated one.
- If an email is provided, a link with the results will be sent when the job is done. The results will be stored for 2 weeks.
Outputs
PRODIGY-CRYSTAL outputs are displayed online and remain downloadable for 14 days. A link to the online resource is also emailed to the user, if an email is provided.
The results returned by the server are divided in four sections and include:
1. PREDICTION RESULTS
- Class of the interface: BIOLOGICAL or CRYSTALLOGRAPHIC
- Probability of the prediction: [this is a number between 0 and 1]
- Protein and chains investigated
- Binding affinity prediction calculated by PRODIGY tools, reported in kcal mol-1
2. PREDICTION DETAILS
- Number of intermolecular residue contacts (RCs) at the interface between the two protein within the threshold distance of 5.0 Å. Contacts are divided by residue-type.
- The Link Density value
- Table of the RCs at the interface, which is a downloadable table (.txt) listing the residues in contact within the given threshold.
3. INTERFACE VISUALIZATION
An online visualization is provided in this section powered by NGL Viewer. The interacting chains are colored in blue/pink for Interface 1 and Interface 2, respectively (see Input part in this Manual). Residues at the interface (i.e., within a threshold of 5.0 Å) are highlighted with green or red surface in the case of biological or crystallographic interfaces, respectively. The complete list of interfacial residues is reported in the Table of the RCs at the interface, reported in the section 2 of the output page.
By moving the mouse on the structure, details of the residue type, residue number, chain ID and atom will appear.
4. DOWNLOAD OUTPUT
In this section is possible to download the PRODIGY-CRYSTAL output folder for your job as archive file, containing all the predictions reported in the online output page.
For more information about the output values and table, check the Method page.